 Dyes & Fluorescence detection chemistry in qPCR
Navarro E, Serrano-Heras G, Castaño MJ, Solera J Clin Chim Acta. 2015 439: 231-250 Real-time
PCR is the method of choice in many laboratories for
diagnostic and food applications. This technology merges the polymerase
chain reaction chemistry with the use of fluorescent reporter molecules
in order to monitor the production of amplification products during
each cycle of the PCR reaction. Thus, the combination of excellent
sensitivity and specificity, reproducible data, low contamination risk
and reduced hand-on time, which make it a post-PCR analysis
unnecessary, has made real-time PCR technology an appealing alternative
to conventional PCR. The present paper attempts to provide a rigorous
overview of fluorescent-based methods for nucleic acid analysis in
real-time PCR described in the literature so far. Herein, different
real-time PCR chemistries have been classified into two main groups;
the first group comprises double-stranded DNA intercalating molecules,
such as SYBR Green I and EvaGreen, whereas the second includes
fluorophore-labeled oligonucleotides. The latter, in turn, has been
divided into three subgroups according to the type of fluorescent
molecules used in the PCR reaction: (i) primer-probes (Scorpions,
Amplifluor, LUX, Cyclicons, Angler); (ii) probes; hydrolysis (TaqMan,
MGB-TaqMan, Snake assay) and hybridization (Hybprobe or FRET, Molecular
Beacons, HyBeacon, MGB-Pleiades, MGB-Eclipse, ResonSense, Yin-Yang or
displacing); and (iii) analogues of nucleic acids (PNA, LNA, ZNA,
non-natural bases: Plexor primer, Tiny-Molecular Beacon). In addition,
structures, mechanisms of action, advantages and applications of such
real-time PCR probes and analogues are depicted in this review.
Randy Rasmussen Ph.D. (Idaho Technology, COO) http://www.idahotec.com/lightcycler_u/lectures/quantification-on-lc.htm SYBR
Green I SYBR Green I is
dsDNA-binding dye. It is thought to bind in the minor groove of
dsDNA and
upon binding increases in fluorescence over a hundred fold (Figure
8a). It is compatible with PCR up to a point, at very high
concentrations
it starts to inhibit the PCR reaction. In the LightCycler
Instrument, SYBR is monitored in channel F1. The biggest advantage of
SYBR is that
it binds to any dsDNA; there is no designing and optimizing of probes
required. If you have a PCR that works, you can have a real-time
quantitative assay working in about a day. The biggest
disadvantage of SYBR is that it binds to any dsDNA; the specific
product, non-specific products and primer dimers are detected equally
well. There are a number of ways to handle this problem.
Careful optimization of the PCR reaction can usually reduce primer
dimers to a level that is only important for very low copy
detection. Hot start techniques like TaqStart antibody can be
helpful in reducing primer dimer. The LightCycler Instrument
allows melting curve analysis of the reaction. This can help to
determine the fraction of the signal coming from the desired product
and the fraction coming from primer dimer. Once the
melting point of the product has been determined the LightCycler
Instrument's flexible programming allows the user to acquire
fluorescence above the melting
temperature of the primer dimers, but below the melting temperature of
the product.
Hybridization Probes If sequence
specific recognition is required, the HybProbe system allows detection
of only the specific product. Two probes are designed that
hybridize side by side on the PCR product (Figure 8c). The 3’ end
of the upstream probe is labeled with fluorescein, which acts as a
fluorescence resonance energy transfer (FRET) donor. The 5’ end
of the downstream probe is labeled with an acceptor dye, either LC Red
640, or LC Red 705. The FRET signal is seen only when two
specific hybridization events occur. In the LightCycler
Instrument, LC Red 640 is monitored in channel F2, LC Red 705 in
channel F3. There may sometimes be an advantage to monitoring the
ration of the acceptor channel (where the signal goes up with
increasing PCR product) and the signal from fluorescein in F1 (which
goes down with increasing PCR product.
TaqMan®
ProbesTaqMan probes
derive their fluorescence signal from the hydrolysis of the probe by
Taq’s 5’ to 3’ exonuclease activity (Figure 8c). The hydrolysis
separates fluorescein from a quenching dye and results in an increased
fluorescein signal. These probes can be used in the LightCycler
Instrument and are monitored in F1 or F1/F2.
The fluorescent
dye SYBR Green I binds to the minor groove of the DNA double helix. In
solution, the unbound dye exhibits very little fluorescence, however,
fluorescence is greatly enhanced upon DNA-binding. Since SYBR Green I
dye is very
stable (only 6% of the activity is lost during 30 amplification cycles)
and the LightCycler instrument's optical filter set matches the
wavelengths of excitation and emission, it is the reagent of choice
when measuring
total DNA. The principle is outlined in the following figures.
At the beginning
of amplification, the reaction mixture contains the denatured DNA, the
primers, and the dye. The unbound dye molecules weakly fluoresce,
producing a minimal background fluorescence signal which is subtracted
during computer analysis.
After annealing
of the
primers, a few dye molecules can bind to the double strand. DNA binding
results
in a dramatic increase of the SYBR Green I molecules to emit light upon
excitation.
During elongation, more and more dye molecules bind to the newly synthesized DNA. If the reaction is monitored continuously, an increase in fluorescence is viewed in real-time. Upon denaturation of the DNA for the next heating cycle, the dye molecules are released and the fluorescence signal falls. Fluorescence
measurement at the end of the elongation step of every PCR cycle is
performed
to monitor the increasing amount of amplified DNA. Together with a
melting curve analysis performed subsequently to the PCR, the SYBR
Green
I format provides an excellent tool for specific product identification
and quantification. Demonstration of preferential binding of SYBR
Green I to specific DNA fragments in real-time multiplex PCR  SYBR Green I (SG) is widely used
in real-time PCR applications as an intercalating dye and is included in many commercially available
kits at undisclosed concentrations. Binding of SG to double-stranded DNA is non-speciÆc and
additional testing, such as DNA melting curve analysis, is required to
conÆrm the generation of a speciÆc
amplicon. The use of melt curve analysis eliminates the necessity for agarose gel electrophoresis
because the melting temperature (Tm) of the speciÆc amplicon is analogous to the detection of an
electrophoretic band. When using SG for real-time PCR multiplex reactions, discrimination of
amplicons should be possible, provided the Tm values are suffiently different. Real-time multiplex
assays for Vibrio cholerae and
Legionella pneumophila using commercially available kits and in-house SG
mastermixes have highlighted variability in performance characteristics, in particular the
detection of only a single
product as assessed by Tm analysis but multiple products as assessed by
agarose gel electrophoresis.
The detected Tm corresponds to the amplicon with the higher G+C%
and larger size, suggesting
preferential binding of SG during PCR and resulting in the failure to
detect multiple amplicons in multiplex reactions when the amount of SG present is limiting. This
has implications for the design and routine application of diagnostic real-time PCR assays employing SG.
A
new minor groove binding asymmetric cyanine reporter dye for real-time
PCR
Martin Bengtsson, H. Jonas Karlsson, Gunnar Westman and Mikael Kubista* Department of Chemistry and Bioscience, Chalmers University of Technology 41296 Go»teborg and TATAA Biocenter, Medicinaregatan 9E, 413 90 Goteborg, Sweden Nucleic Acids Research, 2003, Vol. 31, No. 8 e45 The minor groove binding
asymmetric cyanine dye 4-[(3-methyl-6- (benzothiazol-2-yl)-
2,3-dihydro- (benzo-1,3-thiazole)
-2-methylidene)]- 1-methyl-pyridinium iodide (BEBO) is tested as
sequence nonspeciÆc label in real-time PCR. The
Fluorescence intensity of BEBO increases upon binding to double-stranded DNA allowing
emission to be measured at the end of the elongation phase in the PCR cycle. BEBO concentrations
between 0.1 and 0.4 mM generated sufÆcient Øuorescence
signal without
inhibiting the PCR. A comparison with the commonly used reporter dye
SYBR Green I shows that the two dyes behave similarly
in all important aspects.
BEBO
for qPCR and HRM
TATAA Biocenter
AB, Göteborg, Sweden
BEBO is
an unsymmetric cyanine dye developed by TATAA Biocenter for use in qPCR
applications.
The dye has absorbance and emission wavelengths that can be detected on the FAM channel on most common real-time PCR platforms, and shows a strong fluorescence increase when bound to dsDNA. BEBO can be used as an unspecific dye for real-time PCR applications or other applications where staining of dsDNA is wanted. specific nucleic acid quantification and melting curve analysis. Currently,
in real-time PCR, one often has to choose between using a sequence-specific
probe and a nonspecific double-stranded DNA (dsDNA) binding dye
for the detection of amplified DNA products. The sequence-specific
probe has the advantage that it
only detects the
targeted product, while the nonspecific dye has the
advantage that melting curve analysis can be performed after completed
amplification, which reveals what kind of
products have been formed. Here we present a new
strategy based on combining a sequence-specific probe and a
nonspecific dye, BOXTO, in the same reaction, to take the advantage of
both chemistries.
We show that BOXTO can be used
together with both TaqMan probes and locked nucleic
acid (LNA) probes without interfering with the PCR. The probe signal
reflect formation of target product, while melting curve analysis of
the BOXTO
signal reveals primer-dimer formation and the
presence of any other anomalous products.
BioTechniques - BioSpotlight: Think
outside the BOXTO
Info about BOXTO on the TATAA Biocenter web page BOXTO as a real-time thermal cycling reporter dye ASHRAF I AHMAD Journal of Biosciences, Volume 32, Number 2 / March, 2007 The unsymmetrical cyanine dyes BOXTO (4-[6-(benzoxazole-2-yl-(3-methyl-)-2,3-dihydro- (benzo-1,3-thiazole)-2-methylidene)]- 1-methyl-quinolinium chloride) and its positive divalent derivative BOXTO-PRO (4-[3-methyl-6-(benzoxazole-2-yl)- 2,3-dihydro- (benzo-1,3-thiazole)-2-methylidene)]- 1-(3-trimethylammonium-propyl)- quinolinium dibromide) were studied as real-time PCR reporting fluorescent dyes and compared to SYBR GREEN I (SG) (2-[N-(3-dimethylaminopropyl)-N-propylamino]- 4-[2,3-dihydro-3-methyl- (benzo-1,3-thiazol-2-yl)-methylidene]- 1-phenylquinolinium). Unmodified BOXTO showed no inhibitory effects on real-time PCR, while BOXTO-PRO showed complete inhibition, Sufficient fluorescent signal was acquired when 0.5–1.0 µM BOXTO was used with RotorGene and iCycler platforms. Statistical analysis showed that there is no significant difference between the efficiency and dynamic range of BOXTO and SG. BOXTO stock solution (1.5 mM) was stable at −20°C for more than one year and 40 µM BOXTO solution was more stable than 5x SG when both were stored at 4°C for 45 days. (the ABI TaqMan Probes) Real-time
systems for PCR were improved by probe-based, rather than
intercalator-based, PCR product detection. The principal drawback to
intercalator-based detection of PCR product accumulation is that both
specific and nonspecific products generate signal. An alternative
method, the 5' nuclease assay, provides a real-time method for
detecting only specific amplification products. During amplification,
annealing of the probe to its target sequence generates a substrate
that is cleaved by the 5' nuclease activity of Taq DNA polymerase when
the enzyme extends from an upstream primer into the region of the
probe. This dependence on polymerization ensures that cleavage of the
probe occurs only if the target sequence is being amplified.
The development of fluorogenic probes made it possible to eliminate post-PCR processing for the analysis of probe degradation. The probe is an oligonucleotide with both a reporter fluorescent dye and a quencher dye attached. While the probe is intact, the proximity of the quencher greatly reduces the fluorescence emitted by the reporter dye by Förster resonance energy transfer (FRET) through space. Probe design and synthesis has been simplified by the finding that adequate quenching is observed for probes with the reporter at the 5' end and the quencher at the 3' end. Figure 1 diagrams what happens to a fluorogenic probe during the extension phase of PCR. If the target sequence is present, the probe anneals downstream from one of the primer sites and is cleaved by the 5' nuclease activity of Taq DNA polymerase as this primer is extended. This cleavage of the probe separates the reporter dye from quencher dye, increasing the reporter dye signal. Cleavage removes the probe from the target strand, allowing primer extension to continue to the end of the template strand. Thus, inclusion of the probe does not inhibit the overall PCR process. Additional reporter dye molecules are cleaved from their respective probes with each cycle, effecting an increase in fluorescence intensity proportional to the amount of amplicon produced. The advantage of fluorogenic probes over DNA binding dyes is that specific hybridization between probe and target is required to generate fluorescent signal. Thus, with fluorogenic probes, non-specific amplification due to mis-priming or primer-dimer artifact does not generate signal. Another advantage of fluorogenic probes is that they can be labeled with different, distinguishable reporter dyes. By using probes labeled with different reporters, amplification of two distinct sequences can be detected in a single PCR reaction. The disadvantage of fluorogenic probes is that different probes must be synthesized to detect different sequences: 
The detection principle of LC™ Hybridization Probes (HybProbes) is Fluorescence Resonance Energy Transfer (FRET), the phenomenon of energy transfer from a donor to an acceptor fluorophor. If the donor and the acceptor fluorophor are in close proximity to each other, excitation of the donor by blue light results in energy transfer to the acceptor, which can then emit light of longer wavelength. This fact forms the basis for Roche’s real-time online LightCycler™ PCR System. It allows formation of PCR products to be monitored by using two sequence specific, fluorescent labeled oligonucleotide probes, called Hybridization Probes, in addition to the PCR primers. For this LC™
real-time PCR detection format the following are the major steps:
HybProbes are designed as a pair of which one probe is labeled with the donor (3´Fluo) and one with the acceptor (5´ LCRed 640 or LCRed 705) dye. As FRET decreases with the sixth power of distance, HybProbes have to be designed to hybridise to adjacent regions of the template DNA (separated by 1-5 nucleotides). If both probes hybridise, the two dyes are brought close together and FRET to the acceptor dye results in a signal measurable by the built-in fluorimeter of the LightCycler™. The fluorescence signal disappears by increasing temperature above the melting temperature of the oligos because the probes melt away from the template strand which significantly increases the distance between the dyes. Mismatches
between the
probes and the target decrease the melting temperature of the
respective probe
compared to a perfectly matched probe. This effect can also be used to
detect
SNPs by melting curve analysis.
PCR Monitoring with Hybridization Probes The Hybridization
Probe format is used for DNA detection and quantification and provides
a maximal specificity for product identification. In addition to the
reaction components used for conventional PCR, two specially designed,
sequence specific oligonucleotides labeled with fluorescent dyes are
applied
for this detection method. This allows highly specific detection of the
amplification product as described below.
The first dye (fluorescein) is excited by the LightCycler's LED (Light Emitting Diode) filtered light source, and emits green fluorescent light at a slightly longer wavelength (middle figure). When the two dyes are in close proximity (as shown in the lower figure), the emitted energy excites the LC Red 640 attached to the second hybridization probe that subsequently emits red fluorescent light at an even longer wavelength. This energy transfer, referred to as FRET (Fluorescence Resonance Energy Transfer) is highly dependent on the spacing between the two dye molecules. Only if the molecules are in close proximity (a distance between 1–5 nucleotides) is the energy transferred at high efficiency. Choosing the appropriate detection channel, the intensity of the light emitted by the LightCycler – Red 640 is filtered and measured by the LightCycler instrument's optics. The increasing
amount of measured fluorescence is proportional to the increasing
amount of DNA generated during the ongoing PCR process. Since LC Red
640 only emits a signal when both oligonucleotides are hybridized, the
fluorescence measurement is performed after the annealing step.
Hybridization probes can be labeled with LightCycler – Red 640 and with
LightCycler – Red 705.
The
most difficult qPCR applications demand double-quenched probes for
optimum performance. BHQnova™ probes
have improved quenching efficiency compared to traditional end-labeled
probes, while also improving signal release upon amplification. BHQnova
is most advantageous in longer probe designs, typically those over 25
bases, to boost the signal-to-noise ratio by overcoming the upper limit
on sequence length.
For more product information, visit: our BHQnova Probes Product Info Page The unique
combination of online available assay design software and
only 165 prevalidated, real-time PCR probes allows to quantify
virtually any transcript in the transcriptomes of a large number of
organisms. Universal
ProbeLibrary probes are fully compatible with commonly used
PCR conditions and the hydrolysis probe detection format. They are
labeled at the 5' end with fluorescein (FAM) and at the 3' end with a
dark quencher dye.
Flexibility, specificity, convenience - all in one with the Universal ProbeLibrary The Universal ProbeLibrary combines the flexibility, availability and covenience of SYBR Green I assays with the specificity of Hydrolysis Probe assays. Just 165 prevalidated probes, that can easily be stored in your freezer are sufficient to quantify virtually any transcript from the transcriptomes of a large number of organisms. Target specific intron-spanning qPCR assays are designed online with the ProbeFinder software, freely available at the Universal ProbeLibrary Assay Design Center. The complete assay information, including the sequence of specific primer pairs, and the appropriate Universal ProbeLibrary probe, probe location, amplification product, is displayed on the result page. more info here => Universal ProbeLibrary 
Hybridization Probes for the Detection of Nucleic Acids in Homogeneous Solutions Department of Molecular Genetics, Public Health Research Institute 225 Warren Street, Newark, NJ 07103, USA http://www.molecular-beacons.org/ Table of contents: When
You
Wish Upon A Star: Molecular Beacons: Real Time in a Twinkle Prime
and Shine
While Saving Time: Intergen's Amplifluor allows
Direct Detection of PCR Products
Table of Licensed Providers of
Molecular
Beacons and Kits http://www.synthegen.com/ Specializing in Modified Oligonucleotides
SYNTHEGEN specializes exclusively in modified oligonucleotides.
Differently-colored
molecular probes specific for the wild-type and mutant alleles are
designed. DNA amplified from homozygous wild-type individuals binds
only to the fluorescein-labeled molecular beacons (left). DNA from
homozygous mutants binds only the tetramethylrhodamine-labeled
molecular beacons (right). Both types of molecular probes will bind to
amplicons generated from the DNA of heterozygous individuals
(center).
by L.G.
Kostrikis et al. (1998)
Our genotyping
process is based on Scorpions Technology - a homogeneous or closed tube
method with a simple mix and glow operation. A DNA sample is added to a
Scorpions test and an increase in fluorescence indicates the genotype.
There is no post-PCR manipulation and the use of two fluorescent dyes
gives single tube SNP analysis
Scorpions is a
class leading PCR detection technology with significant benefits
Scorpions
are bi-functional molecules containing a PCR
primer element covalently linked to a probe
element. The molecules also contain a fluorophore that
can interact with a quencher to reduce fluorescence. When the molecules
are
used in a PCR reaction the fluorophore and the quencher are separated
which
leads to an increase in light output from the reaction tube.
The benefits of Scorpions derive from the fact that the probe element is physically coupled to the primer element - this means that the reaction leading to signal generation is a uni-molecular rearrangement. This contrasts to the bi-molecular collisions required by other technologies such as Taqman or Molecular Beacons. The benefits of a
uni-molecular rearrangement are significant - as the reaction is
effectively instantaneous it occurs prior to any competing or side
reactions such as target
amplicon re-annealing or inappropriate target folding. This leads
to stronger signals, more reliable probe design, shorter reaction times
and better discrimination.
The presence of the blocker group is an essential element of the Scorpions invention. Without such a blocker the Taq DNA polymerase would be able to read through the Scorpions primer and copy the probe region. This would generate signal but not in a target specific fashion. Copying the tail in this way would completely negate the benefits of the Scorpions reaction as any inappropriate side-reactions, including the formation of primer dimers, would also generate a signal. Scorpions are PCR
primers with a " Stem-Loop " tail containing a fluorophore and a
quencher
(figue1).
Whitcombe, D., Theaker J., Guy, S.P., Brown, T., Little, S. (1999) Nature 17, 804-807
Molecular
diagnostics is progressing from low-throughput, heterogeneous, mostly
manual technologies to higher throughput, closed-tube, and automated methods.
Fluorescence is the favored signaling technology for such assays,
and a
number of techniques rely on energy transfer between a fluorophore
and a proximal
quencher molecule. In these methods, dual-labeled probes hybridize
to an amplicon and changes in the quenching of
the fluorophore are detected. We describe a new technology that is
simple to use,
gives highly specific information, and avoids the major difficulties of
the alternative methods. It uses a primer with an integral tail
that is used to probe an extension product of the primer. The probing
of a target sequence
is thereby converted into a unimolecular event, which has substantial
benefits in terms of kinetics, thermodynamics, assay design, and probe
reliability.
Design considerations and effects of LNA
in PCR primers
David Latorra1, Khalil Arar*, J. Michael Hurley2 Proligo LLC, 6200 Lookout Road, Boulder, CO 80301, USA Molecular and Cellular Probes 17 (2003) 253–259
The
effects of comprehensive LNA substitution in PCR primers for
amplification of human genomic DNA targets are presented in this
report. Previous research with LNA in other applications has shown
interesting properties for molecular hybridization including enhanced
specificity in allele-specific PCR.
Here we systematically modified PCR primers and conditions for the
human
genomic DNA targets APOB and PAH, along with a b-globin amplification
control, to study whether the number and position of LNA residues
improves
or diminishes amplification sensitivity and specificity. It was
observed
that the design rules for LNA substitution in PCR primers are complex
and depend upon number, position and sequence context. Technical
advantages
were seen when compared to DNA controls for the best LNA primer
designs,
which were typically one to a few centrally located LNA residues. LNA
advantages include increased maximum annealing temperature
ðTmaxÞ
and increased signal with limiting primer or Taq DNA polymerase.
Several
well-characterized designs exhibited different efficiencies with
different
brands of hot-start enzymes. Many shorter LNA primers were found to be
functional compared to same-length non-functional native DNA controls.
These results show that LNA-substituted PCR primers have potential for
use in difficult PCR techniques, such as multiplex amplification at
higher
Tmax; once firm LNA primer design rules are established.
Displacing probes - A new
class of homogeneous nucleic acid probes based on specific displacement
hybridization.
Li Q, Luan G, Guo Q, Liang J. Nucleic Acids Res. 2002 Jan 15;30(2):E5. The Key Laboratory of Cell Biology and Tumor Cell Engineering of the Ministry of Education, Xiamen 361005, Fujian, Chinas
We have developed a new class of
probes for homogeneous nucleic acid detection based on the proposed displacement
hybridization. Our probes consist of two complementary
oligodeoxyribonucleotides of different length labeled with a fluorophore and a quencher in
close proximity in the duplex. The probes on their own are quenched, but they become
fluorescent upon displacement hybridization with the target. These probes
display complete discrimination between a perfectly matched target and
single nucleotide mismatch targets. A comparison of double-stranded probes with
corresponding linear probes confirms that the
presence of the complementary strand significantly enhances their specificity. Using four such probes labeled with different color fluorophores, each designed to recognize a different target, we have demonstrated that multiple targets can be distinguished in the same solution, even if they differ from one another by as little as a single nucleotide. Double-stranded probes were used in real-time nucleic acid amplifications as either probes or as primers. In addition to its extreme specificity and flexibility, the new class of probes is simple to design and synthesize, has low cost and high sensitivity and is accessible to a wide range of labels. This class of probes should find applications in a variety of areas wherever high specificity of nucleic acid hybridization is relevant. Real-time PCR genotyping using
displacing probes.
Cheng J, Zhang Y, Li Q. Nucleic Acids Res. 2004 Apr 15;32(7):e61. The Key Laboratory of Cell Biology and Tumor Cell Engineering of the Ministry of Education, School of Life Sciences, Xiamen University, Xiamen 361005, Fujian, China
Simple and reliable genotyping
technology is a key to success for high-throughput genetic screening
in the post-genome era. Here we have developed a new real-time PCR genotyping
approach that uses displacement hybridization-based probes:
displacing probes. The specificity of displacing probes could be simply assessed
through denaturation analysis before genotyping was implemented, and the probes
designed with maximal specificity also showed the greatest detection
sensitivity. The ease in design, the simple single-dye labeling chemistry and the
capability to adopt degenerated negative strands for point mutation genotyping make the
displacing probes both cost effective and easy to use. The feasibility of
this method was first tested by detecting the C282Y mutation in the human
hemochromatosis gene. The robustness of this approach was then validated by
simultaneous genotyping of five different types of mutation in the human
beta-globin gene. Sixty-two human genomic DNA samples with nine known genotypes were
accurately detected, 32 random clinical samples were successfully screened and 114
double-blind DNA samples were all correctly genotyped. The combined merits of
reliability, flexibility and simplicity should make this method suitable for
routine clinical testing and large-scale genetic screening.
http://www.molbiotech.chalmers.se/research/mk/lightup/lightup.htm Vanvik et al.
developed light-up probes for sequence specific detection of nucleic
acids in homogeneous solution. The probes are made of the nucleic acid
analogue, PNA, and an assymmetric cyanine dye, which upon bind binding
to nucleic acids becomes intesively fluorescent. Under optimum
conditions the probe fluorescence increases 50-fold upon binding to
target DNA and the fluorescence can be observed by the naked eye.
|
||||||||||||||||||||||||||||||||||||||||||||||||


















| |
N. Svanvik, G. Westman, W. Dongyuan & M. Kubista.
Light-up Probes Thiazole Orange Conjugated PNA for Detection of Nucleic
Acid
in Homogeneous Solution. Anal. Biochem. 281, 26-35 (2000). |
| |
N. Svanvik, A. Stålberg, U. Sehlstedt, R. Sjöback
& M. Kubista. Detection of PCR Products in Real-time Using Light-up
Probes. Anal. Biochem. 287, 179-182 (2000). |
| |
N. Svanvik, J. Nygren, G. Westman & M. Kubista.
Free-probe Fluorescence of Light-up probes. J. Amer. Chem. Soc., 123,
803-809 (2001). |
For further information see:

Measure the expression level of any human gene
- Highly sensitive—detect fewer than 10 copies of a DNA target
- Easy multiplex analysis—quantify two or more genes in a single tube
- Accurate over a broad range of target concentrations—detect differences of up to 5 orders of magnitude
- Bright fluorescent signal—works on any qPCR instrument
Introducing
the BD QZyme™ Assay for quantitative PCR (qPCR), a
novel DNA amplification system for the realtime detection and
quantification of specific cDNA and genomic DNA targets. Compatible
with all real-time PCR instruments and readily adapted for use in
single or multiplex analyses, BD QZyme Assays can accurately measure
fewer than 10
copies of target DNA. The assays are easy to set up
and require no optimization since they rely on a
single set of PCR cycling parameters, which can be
universally applied for the detection of any genomic
DNA or mRNA target. The dynamic range, or ability of the assay to
accurately measure differences in target concentration, is
extraordinarily broad, typically extending over
5 orders of magnitude for high-abundance genes.

Amplifluor™ Universal Detection System
http://www.genxpress.at/

Amplifluor Universal Detection System (PDF)
Amplifluor Hous Keeping Direct Gene System (PDF)
Amplifluor Apoptosis Gene System (PDF)
Assay setup for endpoint or real-time protocols
Assay setup for real-time instruments
offer high-performance, cost-effective gene analysis
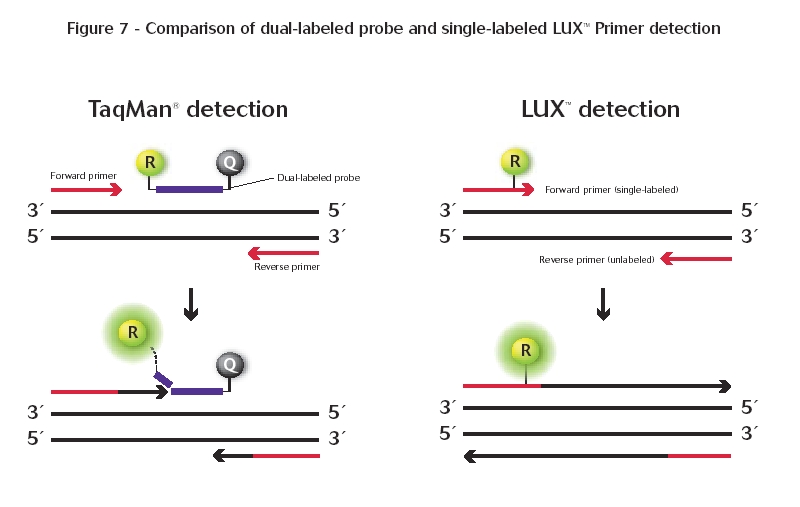
LUX (Light Upon
eXtension) primers

A brochure and
a manual is available on this product
to enable you to
have a better idea of the capacity of our product.
invitrogen-luxprimers-manual.pdf
LUX primers have multiple advantages and among them:
- easy to design: software freely available on Invitrogen website
- multiplexing possibility (advantage over SYBR green)
- melting curve possibility (advantage over TaqMan)
Faye Boeckman, Keith Hamby, and Larissa Tan, Bio-Rad Laboratories, 2000 Alfred Nobel Drive, Hercules, CA 94547 USA
Yuanli Zhang, Dabing Zhang, Wenquan Li, Jianqun Chen, Yufa Peng and Wei Cao
Nucleic Acids Research, 2003, Vol. 31, No. 20 e123